
Research
Mitochondrial complex I plays a critical role in regulating energy generation in cell and it is involved in a number of clinical conditions such as neurodegeneration (Parkinson’s disease, Leber’s optic neuropathy), muscular pathologies and processes of ageing. At the same time ischaemia/reperfusion injury mediated by mitochondria is a major cause of heart disease, as well as stroke; these two are the leading causes of death in the UK, ranking before cancer and causing more than 30% of deaths. Our research is aimed at unraveling the role of mitochondrial complex I and other mitochondrial enzymes in such pathological conditions, translating new findings to clinical medicine.
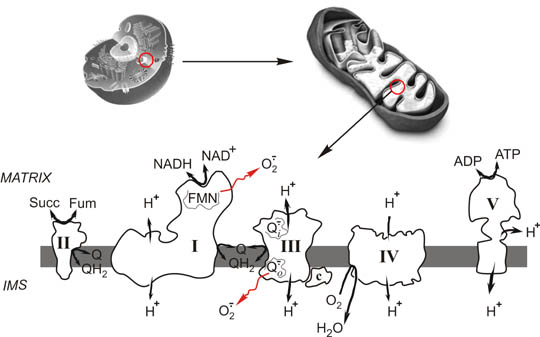
Eukaryotic cell (top, left), mitochondria (top, right) and respiratory chain (bottom). I, Complex I or NADH: ubiquinone oxidoreductase; II, Complex II or succinate dehydrogenase; III, Complex III or bc1 complex or ubiquinol:cytochrome c oxidoreductase; IV, Complex IV or cytochrome c oxidase; Q/QH2 oxidised and reduced membrane ubiquinone, respectively; FMN, tightly bound flavin mononucleotide of Complex I; Succ, succinate; Fum, fumarate; O2 .-, superoxide anion; Qo .- and Qi .-, semiquinones of complex III; H+, protons translocated across the membrane; IMS, intermembrane space.
Hypoxic transition of mitochondrial Complex I
Hypoxia is a lack of oxygen which is sufficient to cause cellular response of the tissue. Hypoxia can be caused by many factors including pathology or trauma, and it also occurs during surgery. Hypoxia is followed by restoration of oxygen supply, so-called reoxygenation, which injures cells and causes potentially irreversible damage. Mitochondria are responsible for such reoxygenation damage during reperfusion, when detrimental oxygen and nitrogen reactive species are formed. Mitochondrial Complex I is a main consumer of the NADH generated in the Krebs cycle as well as a key component of the respiratory chain contributing to the formation of potential across the membrane and therefore to ATP-synthesis. Complex I is known to undergo reversible conformational changes in situations in which its turnover is limited. If the enzyme is idle at physiological temperatures (>30ºC) the mammalian enzyme is rapidly converted into the de-active, dormant form (D-form). This form is catalytically incompetent but can be activated by the slow reaction (k~4 min-1) of NADH oxidation with subsequent ubiquinone reduction. After one or several turnovers the enzyme becomes active (A-form) and can catalyse physiological NADH:ubiquinone reactions at a much higher rate (k~104 min-1). A critical cysteine-39 of the mitochondrially-encoded ND3 subunit has been shown to become exposed on the surface of the D-form and can be specifically labelled by fluorescein-NEM (Fig. 1) [Galkin et al., 2008]. This cysteine residue is not accessible to any covalent modification in the A-form and is responsible for the sensitivity of the D-form to NO-metabolites in isolated mitochondrial membranes and in cultured cells (Fig. 2) [Galkin and Moncada 2007; Galkin et al., 2009].
We demonstrated accumulation of the D-form of complex I in cells after prolonged hypoxia or metabolic hypoxia, when available oxygen cannot be used due to the increase in NO [Galkin et al., 2009]. In such a situation cytochrome c oxidase activity is slowed down and respiratory chain is reduced. Due to the absence of electron acceptor ubiquinone (which is in it’s ubiquinol form), turnover of the enzyme is decreased and Complex I undergoes de-activation. The details of that process are currently under investigation.
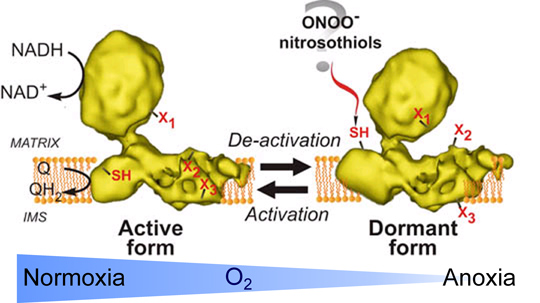
Figure 1. Scheme of A/D transition of Complex I. The active form (A) catalyses the fast NADH:ubiquinone reductase reaction. Absence of substrate results in transformation of the A to the dormant D-form. After conformational re-arrangements during de-activation certain amino acid residues change their position within complex I. Some amino acids, like X1 become buried inside the protein complex. Amino acid residues X2 and X3 become exposed to the outside and susceptible for modification by intracellular effectors, as does the SH-group of cysteine-39 of the mitochondrialy encoded ND3 subunit [Galkin et al., 2008].De-activation of Complex I in hypoxia results in exposure of cysteine-39 of the ND3 subunit which most likely can be modified by S-nitroso-glutathione or peroxynitrite [Galkin&Moncada, 2007; Galkin et al., 2009].
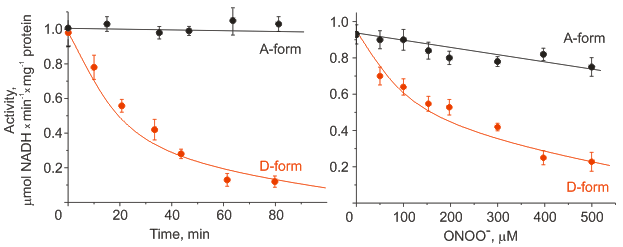
Figure 2. Effect of incubation with 1 mM S-nitrosoglutathione (left) or treatment with peroxynitrite (right) on NADH-oxidase activity of the active and deactive form of Complex I in mitochondrial membranes.
Anaerobic bioenergetics of mitochondrial Complex I
The past decade has revealed a new role for mitochondria in cancer - regulation of oxidative phosphorylation/glycolytic pathways. Prominent features of cancer cells include changes in energy metabolism, mitochondrial alterations and enhanced resistance to apoptosis. The fact that some types of tumor relies heavily on glycolysis and anaerobic respiration to meet their metabolic demands has been recognised by many researchers. Mitochondria of cancer cells associated with hypovascular tumors maintain energy-production in the absence of oxygen. It has been found that some types of cancer cells rely on the mitochondrial NADH:fumarate reductase system to generate energy. The key component of that system is mitochondrial Complex I for which the catalytic properties are not completely understood.
The goals of that project are:
- Characterisation of hypoxic or anaerobic metabolism of cancer cells.
- Identification of the role of hypoxia-induced signalling factors in the regulation of energy metabolism of tumour cells.
Cancer-specific bioenergetics along with changes in the mitochondrial respiratory chain may be taken advantage of for the development of novel classes of anti-tumor agents. Our approach would aim at dissection of energy metabolism of cancer cells by targeting mitochondrial proteins and membranes in response to hypoxia. In our collaborative laboratory, encouraging pre-clinical results have been obtained.
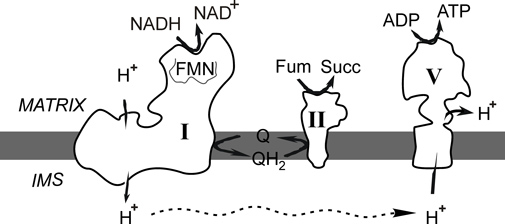
Figure 1. Complex I oxidises matrix NADH and reduces membrane ubiquinone. In the absence of oxygen ubiquinol (QH2) can be oxidised by Complex II reducing fumarate to succinate. The proton gradient formed by Complex I can be used by ATP-synthase to generate ATP.